Who?
- Unit of Lehigh's Library & Technology Services within the Center for Innovation in Teaching & Learning
Our Mission
- We enable Lehigh Faculty, Researchers and Scholars achieve their goals by providing various computational resources; hardware, software, and storage; consulting and training.
Research Computing Staff
- Alex Pacheco, Manager & XSEDE Campus Champion
- Steve Anthony, HPC User Support & System Administrator
- Dan Brashler, CAS Senior Computing Consultant
Introduction to HPC
Library & Technology Services
https://researchcomputing.lehigh.edu
About Us?
What do we do?
- Hardware Support
- Provide system administration and support for Lehigh's HPC clusters.
- 2 University owned and 4 Faculty owned
- Assist with purchase, installation and administration of servers and clusters.
- Provide system administration and support for Lehigh's HPC clusters.
- Data Storage
- Provide data management services including storing and sharing data.
- Software Support
- Provide technical support for software applications, install software as requested and assist with purchase of software.
- Training & Consulting
- Provide education and training programs to facilitate use of HPC resources and general scientific computing needs.
- Provide consultation and support for code development and visualization.
Training & Consulting
- RC staff guest lecture for various courses and provide various training seminars in collaboration with other LTS groups
- Research Computing at Lehigh (Sep. 7, CSE 411)
- Linux: Basic Commands & Environment (Sep. 14, CHM 488, EES 403)
- Using SLURM scheduler on Sol (Sep. 21)
- Shell Scripting (Sep. 28)
- Using Virtualized Software at Lehigh (Oct. 5)
- Python Programming (Oct. 12)
- RefWorks (Oct. 26)
- Document Creation with LaTeX (Nov. 2)
- A Brief Introduction to Linux
- Storage Options at Lehigh
- Research Data Management
- Version Control with GIT
- Programming in MATLAB and GNU Octave
- Enhancing Research Impact
- Programming in R
- Parallel Programming Concepts (ME 413, ACCT 398)
- Saltstack Config Management (CSE 265)
Full Day Workshops
- During the summer we provide full day workshops on programming topics
- Summer 2015 Workshops
- Modern Fortran Programming
- C Programming
- Summer 2017: HPC Parallel Programming Workshop
- Programming in MPI, OpenMP and OpenACC
- We also host full day workshops broadcast from other Supercomputing Centers
- XSEDE HPC Monthly Workshop: OpenACC (Dec. 2014)
- XSEDE HPC Summer BootCamp: OpenMP, OpenACC, MPI and Hybrid Programming (Jun. 2015, 2016 & 2017)
- XSEDE HPC Monthly Workshop: Big Data (Nov. 2015, May 2017 & Dec. 2017)
- To held in EWFM 625 on Dec 5 & 6 from 11AM - 5PM
- Registration at the XSEDE portal: https://portal.xsede.org/course-calendar
Research Computing Resources
Maia
- 32-core Symmetric Multiprocessor (SMP) system available to all Lehigh Faculty, Staff and Students
- dual 16-core AMD Opteron 6380 2.5GHz CPU
- 128GB RAM and 4TB HDD
- Theoretical Performance: 640 GFLOPs (640 billion floating point operations per second)
- Access: Batch Scheduled, no interactive access to Maia
\[ GFLOPs = cores \times clock \times \frac{FLOPs}{cycle} \]
Research Computing Resources
- Sol
- Lehigh's Flagship High Performance Computing Cluster
- 9 nodes, dual 10-core Intel Xeon E5-2650 v3 2.3GHz CPU, 25MB Cache, 128GB RAM, 2x nVIDIA GTX 1080 GPU
- 33 nodes, dual 12-core Intel Xeon E5-2670 v3 2.3Ghz CPU, 30 MB Cache, 128GB RAM, 2x nVIDIA GTX 1080 GPU
- 13 nodes, dual 12-core Intel Xeon E5-2650 v4 2.3Ghz CPU, 30 MB Cache, 64GB RAM
- 1 node, dual 8-core Intel Xeon 2630 v3 2.4GHz CPU, 20 MB Cache, 512GB RAM
- 1TB HDD per node
- 2:1 oversubscribed Infiniband EDR (100Gb/s) interconnect fabric
- Theoretical Performance: 47.37 TFLOPs (CPU) + 28.270 TFLOPs (GPU)
- Access: Batch Scheduled, interactive on login node for compiling, editing only
LTS Managed Faculty Resources
- Monocacy: Ben Felzer, Earth & Environmental Sciences
- Eight nodes, dual 8-core Intel Xeon E5-2650v2, 2.6GHz, 64GB RAM
- Theoretical Performance: 2.662TFlops
- Eight nodes, dual 8-core Intel Xeon E5-2650v2, 2.6GHz, 64GB RAM
- Baltrusaitislab: Jonas Baltrusaitis, Chemical Engineering
- Three nodes, dual 16-core AMD Opteron 6376, 2.3Ghz, 128GB RAM
- Theoretical Performance: 1.766TFlops
- Three nodes, dual 16-core AMD Opteron 6376, 2.3Ghz, 128GB RAM
- Pisces: Keith Moored, Mechanical Engineering and Mechanics
- Six nodes, dual 10-core Intel Xeon E5-2650v3, 2.3GHz, 64GB RAM, nVIDIA Tesla K80
- Theoretical Performance: 4.416 TFlops (CPU) + 17.46TFlops (GPU)
- To be merged with Sol in Fall 2017
- Six nodes, dual 10-core Intel Xeon E5-2650v3, 2.3GHz, 64GB RAM, nVIDIA Tesla K80
- Unnamed : decommissioned faculty cluster for prototyping future resources
- Twenty nodes, dual 8-core Intel Xeon E5-2650v2, 2.6GHz, 64GB RAM
- Theoretical Performance: 6.656TFlops
- Twenty nodes, dual 8-core Intel Xeon E5-2650v2, 2.6GHz, 64GB RAM
Total Computational Resources Supported
| Cluster | Cores | CPU Memory | CPU TFLOPs | GPUs | CUDA Cores | GPU Memory | GPU TFLOPS |
|---|---|---|---|---|---|---|---|
| Maia | 32 | 128 | 0.640 | ||||
| Monocacy | 128 | 512 | 2.662 | ||||
| Unnamed | 320 | 1280 | 6.656 | ||||
| Baltrusaitislab | 96 | 384 | 1.766 | ||||
| Pisces | 120 | 384 | 4.416 | 6 | 29952 | 144 | 17.472 |
| Sol | 1300 | 6720 | 47.366 | 110 | 281600 | 880 | 28.27 |
| Total | 1996 | 9408 | 63.507 | 116 | 311552 | 1024 | 45.742 |
Apply for an account
Apply for an account at the LTS website
- Click on Services > Account & Password > Lehigh Computing Account > Request an account
- Click on the big blue button "Start Special Account Request" > Research Computing Account
- Maia
- Click on "FREE Linux command-line computing"
- Sol: PIs should contact Alex Pacheco or Steve Anthony, web request is not functional
Click on "Fee-based research computing"- Annual charge of $50/account paid by Lehigh Faculty or Research Staff, and
- Annual charge for computing time
Sharing of accounts is explicitly forbidden
Users need to be associated with an allocation to run jobs on Sol
Allocation Charges - Effective Oct. 1, 2016
- Cost per core-hour or service unit (SU) is 1¢
SU is defined as 1 hour of computing on 1 core of the Sol base compute node.
- One base compute node of Sol consumes 20 SU/hour, 480 SU/day and 175,200 SU/year
PIs can share allocations with their collaborators
- Minimum Annual Purchase of 50,000 SU - $500/year
- Additional Increments of 10,000 SU - $100 per 10K increments
- Fixed Allocation cycle: Oct 1 - Sep 30
- Unused allocations do not rollover to next allocation cycle
- Total available computing time for purchase annually: 1.4M SUs or 1 year of continuous computing on 8 nodes
No 'free' computing time provided once allocation has been expended
Condo Investments
- New sustainable model for High Performance Computing at Lehigh
- Faculty (Condo Investor) purchase compute nodes to increase overall capacity of Sol
- LTS will provide for the length of hardware warranty, typically 4 years
- System Administration, Power and Cooling, User Support for Condo Investments
- Condo Investor
- receives annual allocation equivalent to their investment
- can utilize allocations on all available nodes, including nodes from other Condo Investors
- allows idle cycles on investment to be used by other Sol users
- unused allocation will not rollover to the next allocation cycle.
- can purchase additional SUs in 10K increments (minimum 50K not required)
- and must be consumed in current allocation cycle
- Annual Allocation cycle is Oct. 1 - Sep. 30.
Condo Investors
Two at initial launch
- Dimitrios Vavylonis, Physics (1 node)
- Wonpil Im, Biological Sciences (25 nodes, 50 GPUs)
- Anand Jagota, Chemical Engineering (1 node)
- Brian Chen, Computer Science & Engineering (1 node)
- Ed Webb & Alp Oztekin, Mechanical Engineering (6 nodes)
- Jeetain Mittal & Srinivas Rangarajan, Chemical Engineering (13 nodes, 60 GPUs)
- Seth Richards-Shubik, Economics (1 node)
Total SU on Sol after Condo Investments: 11,247,840
Acquisition in progress
- Ganesh Balasubramanian, Mechanical Engineering (at least 10 nodes)
What about Storage resources
- LTS provides various storage options.
- Some of these are in the cloud and subject to Lehigh's Cloud Policy
- For research, LTS provides a 1PB storage system called Ceph
- Ceph is based on the Ceph software
- Research groups can purchase a project space on Ceph @ $200/TB/year that can be shared
- Ceph is in-house, built, operated and administered by LTS Research Computing Staff.
- located in Data Center in EWFM with a backup cluster in Packard Lab
- HPC users can write job output directly to their Ceph volume
- Ceph volume can be mounted as a network drive on Windows or CIFS on Mac and Linux
- See Ceph FAQ for more details
- HPC User home directory quota
- Maia: 5GB
- Sol: 150GB
Network Layout Sol & Ceph Storage Cluster
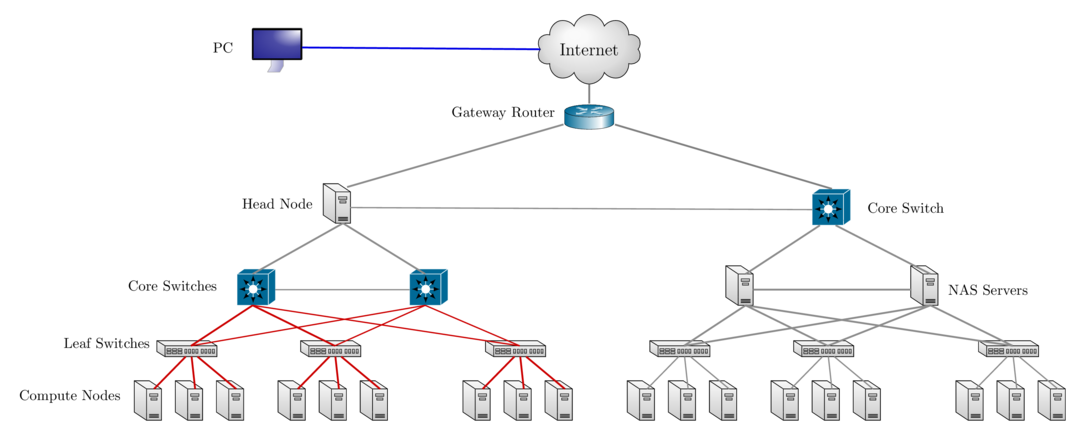
Accessing Research Computing Resources
- Sol & Faculty Clusters: accessible using ssh while on Lehigh's network
ssh username@sol.cc.lehigh.edu
- Maia: No direct access to Maia, instead login to Polaris
ssh username@polaris.cc.lehigh.edu- Polaris is a gateway that also hosts the batch scheduler for Maia
- No computing software including compilers is available on Polaris
- Login to Polaris and request computing time on Maia including interactive access
- On Polaris, run the
maiashellcommand to get interactive access to Maia for 15 minutes.
- On Polaris, run the
- If you are not on Lehigh's network, login to the ssh gateway to get to Research Computing resources
ssh username@ssh.cc.lehigh.edu
Available Software
- Commercial, Free and Open source software is installed on
- Software is managed using module environment
- Why? We may have different versions of same software or software built with different compilers
- Module environment allows you to dynamically change your *nix environment based on software being used
- Standard on many University and national High Performance Computing resource since circa 2011
- How to use Sol/Maia Software on your linux workstation
- LTS provides licensed and open source software for Windows, Mac and Linux and Gogs, a self hosted Git Service or Github clone
Installed Software
- Chemistry/Materials Science
- CPMD
- GAMESS
- Gaussian
- NWCHEM
- Quantum Espresso
- VASP
- Molecular Dynamics
- Desmond
- GROMACS
- LAMMPS
- NAMD
- Computational Fluid Dynamics
- Abaqus
- Ansys
- Comsol
- OpenFOAM
- OpenSees
- Math
- GNU Octave
- Magma
- Maple
- Mathematica
- Matlab
More Software
- Scripting Languages
- R
- Perl
- Python
- Compilers
- GNU
- Intel
- PGI
- CUDA
- Parallel Programming
- MVAPICH2
- OpenMPI
- Libraries
- BLAS/LAPACK/GSL/SCALAPACK
- Boost
- FFTW
- Intel MKL
- HDF5
- NetCDF
- METIS/PARMETIS
- PetSc
- QHull/QRupdate
- SuiteSparse
- SuperLU
More Software
- Visualization Tools
- Avogadro
- GaussView
- GNUPlot
- PWGui
- PyMol
- VMD
- XCrySDen
- Other Tools
- CMake
- Lmod
- Scons
- SPACK
Module Command
| Command | Description |
|---|---|
module avail |
show list of software available on resource |
module load abc |
add software abc to your environment (modify your PATH, LD_LIBRARY_PATH etc as needed) |
module unload abc |
remove abc from your environment |
module swap abc1 abc2 |
swap abc1 with abc2 in your environment |
module purge |
remove all modules from your environment |
module show abc |
display what variables are added or modified in your environment |
module help abc |
display help message for the module abc |
- Users who prefer not to use the module environment will need to modify their
.bashrc or .tcshrc files. Run
module showfor list variables that need modified, appended or prepended
Creating your own modules
- You can always install a software in your home directory
- Stay compliant with software licensing
- Modify your .bashrc/.tcshrc to add software to your path, OR
- create a module and dynamically load it so that it doesn't interfere
with other software installed on the system
- e.g. You might want a different version of openmpi installed
- the system admin may not want install it system wide for just one user
- Add the directory where you will install the module files to the variable MODULEPATH in .bashrc/.tcshrc
# My .bashrc file
export MODULEPATH=${MODULEPATH}:/home/alp514/modulefiles
Module File Example
Compilers
- Various versions of compilers installed on Sol
- Open Source: GNU Compiler (also called gcc even though gcc is the c compiler)
- 4.8.5 (system default), 5.3.0, 6.1.0 and 7.1.0
- Commercial: Only two seats of each
- Intel Compiler: 16.0.3, 17.0.0 and 17.0.3
- Portland Group or PGI: 16.5, 16.10, 17.4 and 17.7
- We are licensed to install any available version
- On Sol, all except gcc 4.8.5 are available via the module environment
| Language | GNU | Intel | PGI |
|---|---|---|---|
| Fortran | gfortran | ifort | pgfortran |
| C | gcc | icc | pgcc |
| C++ | g++ | icpc | pgc++ |
Compiling Code
- Usage:
<compiler> <options> <source code> - Example:
ifort -o saxpyf saxpy.f90gcc -o saxpyc saxpy.c
- Common Compiler options or flags
-o myexec: compile code and create an executable myexec. If this option is not given, then a defaulta.outis created.-l{libname}: link compiled code to a library called libname. e.g. to use lapack libraries, add-llapackas a compiler flag.-L{directory path}: directory to search for libraries. e.g.-L/usr/lib64 -llapackwill search for lapack libraries in /usr/lib64.-I{directory path}: directory to search for include files and fortran modules.
Compilers for Parallel Programming: OpenMP & TBB
- OpenMP support is built-in
| Compiler | OpenMP Flag | TBB Flag |
|---|---|---|
| GNU | -fopenmp | -L$TBBROOT/lib/intel64_lin/gcc4.4 -ltbb |
| Intel | -qopenmp | -L$TBBROOT/lib/intel64_lin/gcc4.4 -ltbb |
| PGI | -mp |
- TBB is available as part of Intel Compiler suite
$TBBROOTdepends on the Intel Compiler Suite you want to use.- Not sure if this will work for PGI Compilers
[alp514.sol](1083): module show intel
-------------------------------------------------------------------
/share/Apps/share/Modules/modulefiles/toolchain/intel/16.0.3:
module-whatis Set up Intel 16.0.3 compilers.
conflict pgi
conflict gcc
setenv INTEL_LICENSE_FILE /share/Apps/intel/licenses/server.lic
setenv IPPROOT /share/Apps/intel/compilers_and_libraries_2016.3.210/linux/ipp
setenv MKLROOT /share/Apps/intel/compilers_and_libraries_2016.3.210/linux/mkl
setenv TBBROOT /share/Apps/intel/compilers_and_libraries_2016.3.210/linux/tbb
...
snip
...
Compilers for Parallel Programming: MPI
- MPI is a library and not a compiler, built or compiled for different compilers.
| Language | Compile Command |
|---|---|
| Fortran | mpif90 |
| C | mpicc |
| C++ | mpicxx |
- Usage:
<compiler> <options> <source code>
[2017-10-30 08:40.30] ~/Workshop/2017XSEDEBootCamp/MPI/Solutions
[alp514.sol](1096): mpif90 -o laplace_f90 laplace_mpi.f90
[2017-10-30 08:40.45] ~/Workshop/2017XSEDEBootCamp/MPI/Solutions
[alp514.sol](1097): mpicc -o laplace_c laplace_mpi.c
[2017-10-30 08:40.57] ~/Workshop/2017XSEDEBootCamp/MPI/Solutions
- The MPI compiler command is just a wrapper around the underlying compiler
[alp514.sol](1080): mpif90 -show
ifort -fPIC -I/share/Apps/mvapich2/2.1/intel-16.0.3/include
-I/share/Apps/mvapich2/2.1/intel-16.0.3/include
-L/share/Apps/mvapich2/2.1/intel-16.0.3/lib
-lmpifort -Wl,-rpath -Wl,/share/Apps/mvapich2/2.1/intel-16.0.3/lib
-Wl,--enable-new-dtags -lmpi
MPI Libraries
- There are two different MPI implementations commonly used
MPICH: Developed by Argonned National Laboratory- used as a starting point for various commercial and open source MPI libraries
MVAPICH2: Developed by D. K. Panda with support for InfiniBand, iWARP, RoCE, and Intel Omni-Path. (default MPI on Sol)Intel MPI: Intel's version of MPI. You need this for Xeon Phi MICs.- available in cluster edition of Intel Compiler Suite. Not available at Lehigh
IBM MPIfor IBM BlueGene andCRAY MPIfor Cray systems
OpenMPI: A Free, Open Source implementation from merger of three well know MPI implementations. Can be used for commodity network as well as high speed networkFT-MPIfrom the University of TennesseeLA-MPIfrom Los Alamos National LaboratoryLAM/MPIfrom Indiana University
Running MPI Programs
- Every MPI implementation come with their own job launcher:
mpiexec(MPICH,OpenMPI & MVAPICH2),mpirun(OpenMPI) ormpirun_rsh(MVAPICH2) - Example:
mpiexec [options] <program name> [program options] - Required options: number of processes and list of hosts on which to run program
| Option Description | mpiexec | mpirun | mpirun_rsh |
|---|---|---|---|
run on x cores |
-n x | -np x | -n x |
| location of the hostfile | -f filename | -machinefile filename | -hostfile filename |
- To run a MPI code, you need to use the launcher from the same implementation that was used to compile the code.
- For e.g.: You cannot compile code with OpenMPI and run using the MPICH and MVAPICH2's launcher
- Since MVAPICH2 is based on MPICH, you can launch MVAPICH2 compiled code using MPICH's launcher.
- SLURM scheduler provides
srunas a wrapper around all mpi launchers
Running MPI Codes
[2017-10-30 08:47.46] ~/Workshop/2017XSEDEBootCamp/MPI/Solutions
[alp514.sol-b112](993): mpiexec -n 4 ./laplace_f90
Maximum iterations [100-4000]?
200
---------- Iteration number: 100 ---------------
( 995, 995): 63.33 ( 996, 996): 72.67 ( 997, 997): 81.40 ( 998, 998): 88.97 ( 999, 999): 94.86 (1000,1000): 98.67
Max error at iteration 200 was 0.177397842364442
Total time was 0.1172750 seconds.
---------- Iteration number: 200 ---------------
( 995, 995): 79.11 ( 996, 996): 84.86 ( 997, 997): 89.91 ( 998, 998): 94.10 ( 999, 999): 97.26 (1000,1000): 99.28
[2017-10-30 08:47.57] ~/Workshop/2017XSEDEBootCamp/MPI/Solutions
[alp514.sol-b112](994): mpiexec -n 4 ./laplace_c
Maximum iterations [100-4000]?
200
---------- Iteration number: 100 ------------
[995,995]: 63.33 [996,996]: 72.67 [997,997]: 81.40 [998,998]: 88.97 [999,999]: 94.86 [1000,1000]: 98.67
---------- Iteration number: 200 ------------
[995,995]: 79.11 [996,996]: 84.86 [997,997]: 89.91 [998,998]: 94.10 [999,999]: 97.26 [1000,1000]: 99.28
Max error at iteration 200 was 0.177398
Total time was 0.259370 seconds.
[2017-10-30 08:49.05] ~
[alp514.sol-b112](1000): srun $(which lammps) -in in.lj -var n 1 -var x 1
LAMMPS (14 May 2016)
Lattice spacing in x,y,z = 1.6796 1.6796 1.6796
Created orthogonal box = (0 0 0) to (33.5919 1.6796 1.6796)
24 by 1 by 1 MPI processor grid
... snipped ...
Compiling Using Makefiles
- Makefile is a file containing a set of directives used with the make build automation tool.
- directs make on how to compile and link a program.
- Using C/C++ as an example, when a C/C++ source file is changed, it must be recompiled.
- If a header file has changed, each C/C++ source file that includes the header file must be recompiled to be safe.
- Each compilation produces an object file corresponding to the source file.
- Finally, if any source file has been recompiled, all the object files, whether newly made or saved from previous compilations, must be linked together to produce the new executable program.
- These instructions with their dependencies are specified in a makefile.
- If none of the files that are prerequisites have been changed since the last time the program was compiled, no actions take place.
- For large software projects, using Makefiles can substantially reduce build times if only a few source files have changed.
Makefile Examples
ifeq ($(COMP),gnu)
CC = gcc
FC = gfortran
CFLAGS = -cpp
OFLAGS = -fopenmp
BINF = pif pif_omp pif_ompr
BINC = pic pic_omp pic_ompr
else ifeq ($(COMP),intel)
CC = icc
FC = ifort
CFLAGS = -fpp
OFLAGS = -qopenmp
BINF = pif pif_omp pif_ompr
BINC = pic pic_omp pic_ompr
else
CC = pgcc
FC = pgf90
CFLAGS = -Mpreprocess
OFLAGS = -mp
AFLAGS = -acc -Minfo=accel -ta=tesla:cc60 -Mcuda=kepler+
BINF = pif pif_omp pif_ompr pif_acc
BINC = pic pic_omp pic_ompr pic_acc
endif
ifeq ($(precision),single)
PREC = -Mpreprocess
else
PREC = -DDP -Mpreprocess
endif
all: $(BINC) $(BINF)
pic:
$(CC) $(CFLAGS) $(PREC) -o pic pi_serial.c
pif:
$(FC) $(CFLAGS) $(PREC) -o pif pi_serial.f90
pic_ompr:
$(CC) $(CFLAGS) $(PREC) $(OFLAGS) -o pic_ompr pi_ompr.c
pic_omp:
$(CC) $(CFLAGS) $(PREC) $(OFLAGS) -o pic_omp pi_omp.c
pif_ompr:
$(FC) $(CFLAGS) $(PREC) $(OFLAGS) -o pif_ompr pi_ompr.f90
pif_omp:
$(FC) $(CFLAGS) $(PREC) $(OFLAGS) -o pif_omp pi_omp.f90
pic_acc:
$(CC) $(CFLAGS) $(PREC) $(AFLAGS) -o pic_acc pi_acc.c
pif_acc:
$(FC) $(CFLAGS) $(PREC) $(AFLAGS) -o pif_acc pi_acc.f90
clean:
rm -rf $(BINC) $(BINF) *~
Cluster Environment
- A cluster is a group of computers (nodes) that works together closely
Two types of nodes
- Head/Login Node
- Compute Node
Multi-user environment
Each user may have multiple jobs running simultaneously
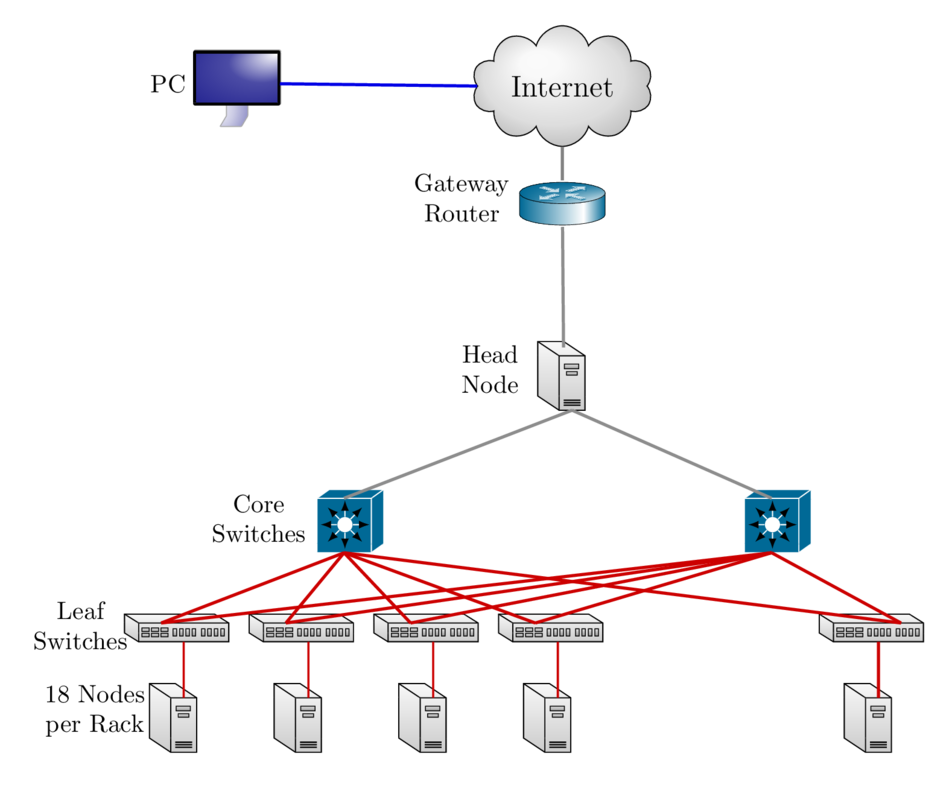
How to run jobs
- All compute intensive jobs are scheduled
- Write a script to submit jobs to a scheduler
- need to have some background in shell scripting (bash/tcsh)
- Need to specify
- Resources required (which depends on configuration)
- number of nodes
- number of processes per node
- memory per node
- How long do you want the resources
- have an estimate for how long your job will run
- Which queue to submit jobs
- SLURM uses the term partition instead of queue
- Resources required (which depends on configuration)
Scheduler & Resource Management
A software that manages resources (CPU time, memory, etc) and schedules job execution
- Sol: Simple Linux Utility for Resource Management (SLURM)
- Others: Portable Batch System (PBS)
- Scheduler: Maui
- Resource Manager: Torque
- Allocation Manager: Gold
A job can be considered as a user’s request to use a certain amount of resources for a certain amount of time
The Scheduler or queuing system determines
- The order jobs are executed
- On which node(s) jobs are executed
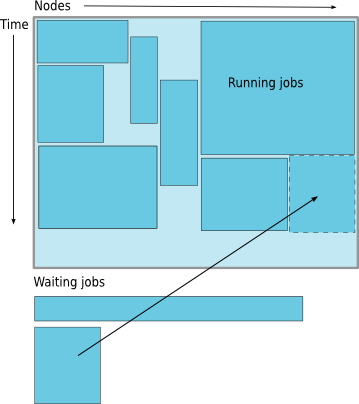
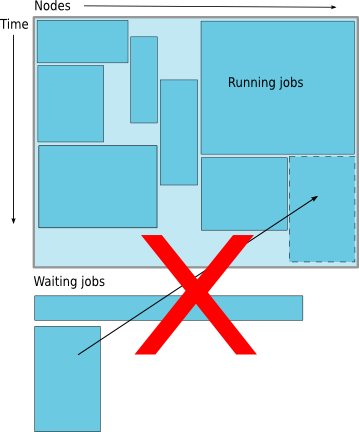
Job Scheduling
Map jobs onto the node-time space
- Assuming CPU time is the only resource
Need to find a balance between
- Honoring the order in which jobs are received
- Maximizing resource utilization
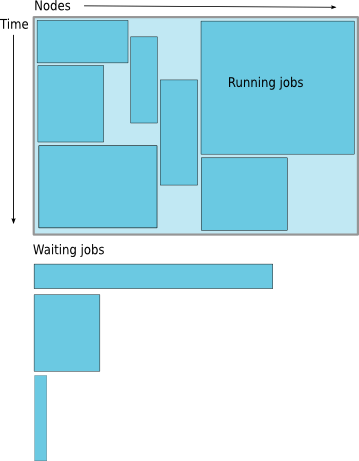
Backfilling
- A strategy to improve utilization
- Allow a job to jump ahead of others when there are enough idle nodes
- Must not affect the estimated start time of the job with the highest priority
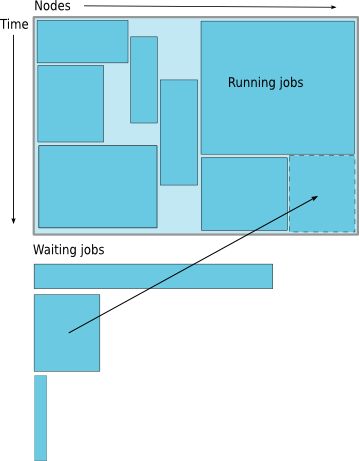
How much time must I request
- Ask for an amount of time that is
- Long enough for your job to complete
- As short as possible to increase the chance of backfilling
Available Queues
| Cluster | Partition Name | Max Runtime in hours | Max SU consumed node per hour |
|---|---|---|---|
| Sol | lts | 72 | 20 (will change to 18+2) |
| imlab | 48 | 22 | |
| imlab-gpu | 48 | 24 | |
| eng | 72 | 24 (will change to 22+2) | |
| engc | 72 | 24 (will change to 22+2) | |
| all-cpu | 48 | ||
| all-gpu | 48 |
- Maia
| Queue Name | Max Runtime in hours | Max Simultaneous Core-hours |
|---|---|---|
| smp-test | 1 | 4 |
| smp | 96 | 384 |
How much memory can or should I use per core?
The amount of installed memory less the amount that is used by the operating system and other utilities
A general rule of thumb on most HPC resources: leave 1-2GB for the OS to run.
| Cluster | Partition | Max Memory/core (GB) | Recommended Memory/Core (GB) |
|---|---|---|---|
| Sol | lts | 6.4 | 6.2 |
| eng/imlab/imlab-gpu | 5.3 | 5.1 | |
| engc | 2.66 | 2.4 |
if you need to run a single core job that requires 10GB memory in the imlab partition, you need to request 2 cores even though you are only using 1 core.
Maia: Users need to specify memory required in their submit script. Max memory that should be requested is 126GB.
Useful SBATCH Directives
| SLURM Directive | Description |
|---|---|
| #SBATCH --partition=queuename | Submit job to the queuename partition. |
| #SBATCH --time=hh:mm:ss | Request resources to run job for hh hours, mm minutes and ss seconds. |
| #SBATCH --nodes=m | Request resources to run job on m nodes. |
| #SBATCH --ntasks-per-node=n | Request resources to run job on n processors on each node requested. |
| #SBATCH --ntasks=n | Request resources to run job on a total of n processors. |
| #SBATCH --job-name=jobname | Provide a name, jobname to your job. |
| #SBATCH --output=filename.out | Write SLURM standard output to file filename.out. |
| #SBATCH --error=filename.err | Write SLURM standard error to file filename.err. |
| #SBATCH --mail-type=events | Send an email after job status events is reached. |
| events can be NONE, BEGIN, END, FAIL, REQUEUE, ALL, TIME_LIMIT(_90,80) | |
| #SBATCH --mail-user=address | Address to send email. |
| #SBATCH --account=mypi | charge job to the mypi account |
Useful SBATCH Directives (contd)
| SLURM Directive | Description |
|---|---|
| #SBATCH --qos=nogpu | Request a quality of service (qos) for the job in imlab, engc partitions. |
| Job will remain in queue indefinitely if you do not specify qos | |
| #SBATCH --gres=gpu:# | Specifies number of gpus requested in the gpu partitions |
| You can request 1 or 2 gpus with a minimum of 1 core or cpu per gpu |
- SLURM can also take short hand notation for the directives
| Long Form | Short Form |
|---|---|
| --partition=queuename | -p queuename |
| --time=hh:mm:ss | -t hh:mm:ss |
| --nodes=m | -N m |
| --ntasks=n | -n n |
| --account=mypi | -A mypi |
| --job-name=jobname | -J jobname |
| --output=filename.out | -o filename.out |
SBATCH Filename Patterns
- sbatch allows for a filename pattern to contain one or more replacement symbols, which are a percent sign "%" followed by a letter (e.g. %j).
| Pattern | Description |
|---|---|
| %A | Job array's master job allocation number. |
| %a | Job array ID (index) number. |
| %J | jobid.stepid of the running job. (e.g. "128.0") |
| %j | jobid of the running job. |
| %N | short hostname. This will create a separate IO file per node. |
| %n | Node identifier relative to current job (e.g. "0" is the first node of the running job) This will create a separate IO file per node. |
| %s | stepid of the running job. |
| %t | task identifier (rank) relative to current job. This will create a separate IO file per task. |
| %u | User name. |
| %x | Job name. |
Useful PBS Directives
| PBS Directive | Description |
|---|---|
| #PBS -q queuename | Submit job to the queuename queue. |
| #PBS -l walltime=hh:mm:ss | Request resources to run job for hh hours, mm minutes and ss seconds. |
| #PBS -l nodes=m:ppn=n | Request resources to run job on n processors each on m nodes. |
| #PBS -l mem=xGB | Request xGB per node requested, applicable on Maia only |
| #PBS -N jobname | Provide a name, jobname to your job. |
| #PBS -o filename.out | Write PBS standard output to file filename.out. |
| #PBS -e filename.err | Write PBS standard error to file filename.err. |
| #PBS -j oe | Combine PBS standard output and error to the same file. |
| #PBS -M your email address | Address to send email. |
| #PBS -m status | Send an email after job status status is reached. |
| status can be a (abort), b (begin) or e (end). The arguments can be combined | |
| for e.g. abe will send email when job begins and either aborts or ends |
Useful PBS/SLURM environmental variables
| SLURM Command | Description | PBS Command |
|---|---|---|
| SLURM_SUBMIT_DIR | Directory where the qsub command was executed |
PBS_O_WORKDIR |
| SLURM_JOB_NODELIST | Name of the file that contains a list of the HOSTS provided for the job | PBS_NODEFILE |
| SLURM_NTASKS | Total number of cores for job | PBS_NP |
| SLURM_JOBID | Job ID number given to this job | PBS_JOBID |
| SLURM_JOB_PARTITION | Queue job is running in | PBS_QUEUE |
| Walltime in secs requested | PBS_WALLTIME | |
| Name of the job. This can be set using the -N option in the PBS script | PBS_JOBNAME | |
| Indicates job type, PBS_BATCH or PBS_INTERACTIVE | PBS_ENVIRONMENT | |
| value of the SHELL variable in the environment in which qsub was executed | PBS_O_SHELL | |
| Home directory of the user running qsub | PBS_O_HOME |
Basic Job Manager Commands
- Submission
- Monitoring
- Manipulating
- Reporting
Job Types
- Interactive Jobs
- Set up an interactive environment on compute nodes for users
- Will log you into a compute node and wait for your prompt
- Purpose: testing and debugging code. Do not run jobs on head node!!!
- All compute node have a naming convention sol-[a,b,c]###
- head node is sol
- Batch Jobs
- Executed using a batch script without user intervention
- Advantage: system takes care of running the job
- Disadvantage: cannot change sequence of commands after submission
- Useful for Production runs
- Workflow: write a script -> submit script -> take mini vacation -> analyze results
- Executed using a batch script without user intervention
Job Types: Interactive
PBS: Use
qsub -Icommand with PBS Directives-
qsub -I -V -l walltime=<hh:mm:ss>,nodes=<# of nodes>:ppn=<# of core/node> -q <queue name>
-
SLURM: Use
sruncommand with SBATCH Directives followed by--pty /bin/bashsrun --time=<hh:mm:ss> --nodes=<# of nodes> --ntasks-per-node=<# of core/node> -p <queue name> --pty /bin/bash- If you have
soltoolsmodule loaded, then useinteractwith at least one SBATCH Directiveinteract -t 20[Assumes-p lts -n 1 -N 20]
Run a job interactively replace
--pty /bin/bash --loginwith the appropriate command.- For e.g.
srun -t 20 -n 1 -p imlab --qos=nogpu $(which lammps) -in in.lj -var x 1 -var n 1 - Default values are 3 days, 1 node, 20 tasks per node and lts partition
- For e.g.
Job Types: Batch
- Workflow: write a script -> submit script -> take mini vacation -> analyze results
Submitting Batch Jobs
- PBS:
qsub filename - SLURM:
sbatch filename
- PBS:
qsubandsbatchcan take the options for#PBSand#SBATCHas command line argumentsqsub -l walltime=1:00:00,nodes=1:ppn=16 -q normal filenamesbatch --time=1:00:00 --nodes=1 --ntasks-per-node=20 -p lts filename
Minimal submit script for Serial Jobs
#!/bin/bash
#PBS -q smp
#PBS -l walltime=1:00:00
#PBS -l nodes=1:ppn=1
#PBS -l mem=4GB
#PBS -N myjob
cd ${PBS_O_WORKDIR}
./myjob < filename.in > filename.out
#!/bin/bash
#SBATCH --partition=lts
#SBATCH --time=1:00:00
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --job-name myjob
cd ${SLURM_SUBMIT_DIR}
./myjob < filename.in > filename.out
Minimal submit script for MPI Job
#!/bin/bash
#SBATCH --partition=lts
#SBATCH --time=1:00:00
#SBATCH --nodes=2
#SBATCH --ntasks-per-node=20
## For --partition=imlab,
### use --ntasks-per-node=22
### and --qos=nogpu
#SBATCH --job-name myjob
module load mvapich2
cd ${SLURM_SUBMIT_DIR}
srun ./myjob < filename.in > filename.out
exit
#!/bin/bash
#PBS -q normal
#PBS -l walltime=1:00:00
#PBS -l nodes=2:ppn=16
#PBS -N myjob
module load openmpi/1.6.5/pgi/13.10
cd ${PBS_O_WORKDIR}
mpiexec -n $PBS_PPN -f $PBS_NODEFILE ./myjob \
< filename.in > filename.out
exit
Minimal submit script for OpenMP Job
#!/bin/tcsh
#SBATCH --partition=imlab
# Directives can be combined on one line
#SBATCH --time=1:00:00 --nodes=1 --ntasks-per-node=22
#SBATCH --qos=nogpu
#SBATCH --job-name myjob
cd ${SLURM_SUBMIT_DIR}
# Use either
setenv OMP_NUM_THREADS 22
./myjob < filename.in > filename.out
# OR
OMP_NUM_THREADS=22 ./myjob < filename.in > filename.out
exit
#!/bin/bash
#PBS -q normal
#PBS -l walltime=1:00:00
#PBS -l nodes=1:ppn=32
#PBS -N myjob
cd ${PBS_O_WORKDIR}
export OMP_NUM_THREADS=32
./myjob < filename.in > filename.out
exit
Minimal submit script for LAMMPS GPU job
#!/bin/tcsh
#SBATCH --partition=imlab
# Directives can be combined on one line
#SBATCH --time=1:00:00
#SBATCH --nodes=1
# 1 CPU can be be paired with only 1 GPU
# 1 GPU can be paired with all 24 CPUs
#SBATCH --ntasks-per-node=1
#SBATCH --gres=gpu:1
# Need both GPUs, use --gres=gpu:2
#SBATCH --job-name myjob
cd ${SLURM_SUBMIT_DIR}
# Load LAMMPS Module
module load lammps/17nov16-gpu
# Run LAMMPS for input file in.lj
srun $(which lammps) -in in.lj -sf gpu -pk gpu 1 gpuID ${CUDA_VISIBLE_DEVICE}
exit
Monitoring & Manipulating Jobs
| SLURM Command | Description | PBS Command |
|---|---|---|
| squeue | check job status (all jobs) | qstat |
| squeue -u username | check job status of user username | qstat -u username |
| squeue --start | Show estimated start time of jobs in queue | showstart jobid |
| scontrol show job jobid | Check status of your job identified by jobid | checkjob jobid |
| scancel jobid | Cancel your job identified by jobid | qdel jobid |
| scontrol hold jobid | Put your job identified by jobid on hold | qhold jobid |
| scontrol release jobid | Release the hold that you put on jobid | qrls jobid |
- The following scripts written by RC staff can also be used for monitoring jobs.
- checkq:
squeuewith additional useful option. - checkload:
sinfowith additional options to show load on compute nodes.
- checkq:
- load the
soltoolsmodule to get access to RC staff created scripts
Usage Reporting
- sacct: displays accounting data for all jobs and job steps in the SLURM job accounting log or Slurm database
sshare: Tool for listing the shares of associations to a cluster.
We have created scripts based on these to provide usage reporting
-
alloc_summary.sh- included in your .bash_profile
- prints allocation usage on your login shell
-
balance- prints allocation usage summary
-
solreport- obtain your monthly usage report
- PIs can obtain usage report for all or specific users on their allocation
- use
--helpfor usage information
-
Need to run multiple jobs in sequence?
- Option 1: Submit jobs as soon as previous jobs complete
Option 2: Submit jobs with a dependency
You want to run several serial processor jobs on
- one node: your submit script should be able to run several serial
jobs in background and then use the
waitcommand for all jobs to finish - more than one node: this requires some background in scripting but the idea is the same as above
- one node: your submit script should be able to run several serial
jobs in background and then use the
Additional Help & Information
- Issue with running jobs or need help to get started:
- Open a help ticket: http://go.lehigh.edu/rchelp
- More Information
- Subscribe
- HPC Training Google Groups: hpctraining-list+subscribe@lehigh.edu
- Research Computing Mailing List: https://lists.lehigh.edu/mailman/listinfo/hpc-l
- My contact info
- eMail: alp514@lehigh.edu
- Tel: (610) 758-6735
- Location: Room 296, EWFM Computing Center
- My Schedule